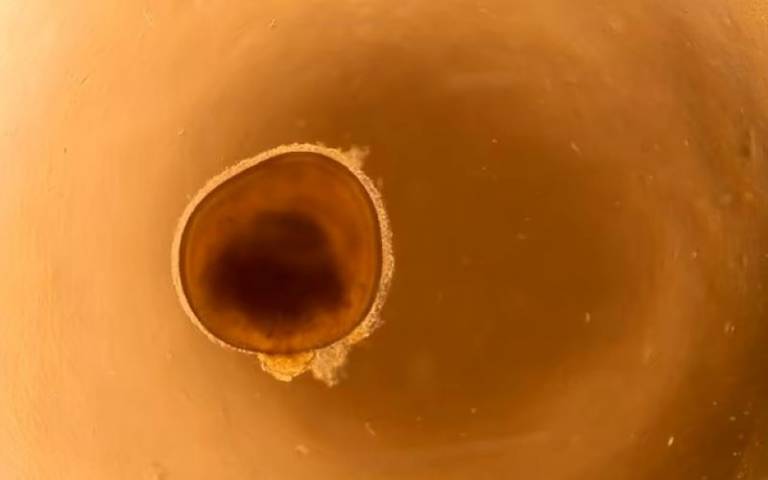
Lab-Grown Retina Uncovers Gene Clue for Rare Childhood Eye Disease
For families navigating the complexities of a rare genetic condition, answers can feel frustratingly out of reach. This is especially true for Leber congenital amaurosis (LCA), a group of inherited retinal diseases that cause severe vision loss from infancy. But now, a groundbreaking approach using lab-grown human retinal tissue has illuminated a previously unknown genetic mechanism behind one form of this condition, offering new hope for future therapies.
Scientists have successfully used these intricate retinal “organoids” to model a specific type of LCA linked to mutations in the NPHP5 gene. Their discoveries, published in the journal *Stem Cell Reports*, not only reveal why these mutations cause blindness but also point to a potential pathway for intervention.
What is Leber Congenital Amaurosis (LCA)?
Leber congenital amaurosis is a devastating diagnosis. It is one of the most common causes of inherited blindness in children, affecting approximately 1 in 40,000 newborns. Children with LCA typically have:
- Severely impaired vision or blindness at birth or within the first few months of life.
- Nystagmus, or involuntary “wobbly” eye movements.
- Poor pupillary response to light.
- Deeply sunken eyes, a symptom known as enophthalmos.
The condition is caused by mutations in at least 25 different genes, all of which are critical for the function and survival of photoreceptor cells—the rods and cones in the retina that convert light into electrical signals for the brain. The specific type studied here, NPHP5-associated LCA, also frequently leads to kidney failure later in life, a condition known as Senior-Løken syndrome.
The Power of a Miniature Retina Grown in a Lab
Traditional research into such rare diseases faces significant hurdles. Animal models don’t always perfectly mimic human biology, and studying developing human retinal tissue directly is ethically and practically impossible. This is where retinal organoids change the game.
Researchers start with human skin or blood cells, reprogramming them back into induced pluripotent stem cells (iPSCs). These “master cells” are then coaxed with specific chemical signals to develop into a complex, three-dimensional tissue that remarkably resembles the architecture of a human retina. It forms layers of different cell types, including the crucial photoreceptors.
Modeling Disease in a Dish
For this study, scientists used gene-editing technology on stem cells to create two models:
- A line with a specific NPHP5 mutation known to cause LCA in patients.
- A “corrected” line where the mutation was fixed, serving as a healthy control.
They then grew retinal organoids from both sets of cells and watched as they developed over months, comparing the diseased and healthy tissues side-by-side. This allowed them to witness, for the first time in human tissue, exactly what goes wrong during development.
The Critical Discovery: A Cilia Connection
The NPHP5 protein is known to be involved in the function of primary cilia, tiny hair-like structures on the surface of cells that act as critical signaling hubs. In photoreceptors, a specialized type of cilium forms the connecting cilium, which is the essential lifeline between the inner cell body and the light-sensing outer segment.
The lab-grown retina revealed the precise defect:
- In the organoids with the NPHP5 mutation, the photoreceptor cells initially developed normally and began forming their light-sensing outer segments.
- However, these outer segments then failed to mature and stabilize. They became disorganized and started to degenerate.
- This degeneration was linked to a severe disruption in the transport of proteins along the connecting cilium. Key proteins like rhodopsin, vital for detecting light, could not make their journey to the outer segment.
- Ultimately, without a functional outer segment, the photoreceptors underwent programmed cell death (apoptosis), leading to blindness.
“This was a key finding,” explained a senior researcher on the project. “The organoids showed us that the problem isn’t that the photoreceptors never form—it’s that they cannot maintain the intricate structure they need to sense light. The connecting cilium is like a broken supply chain, and the cell factory shuts down.”
From Discovery to Future Treatment Pathways
Perhaps the most exciting part of this research is that it didn’t just diagnose the problem; it also hinted at a solution. The study identified a specific molecular pathway that was downregulated because of the ciliary defect.
A Potential Pharmacological Fix
When researchers treated the diseased retinal organoids with a small molecule drug designed to boost the activity of this pathway, they observed a promising effect: the photoreceptor cells showed reduced signs of stress and degeneration.
This suggests that NPHP5-associated LCA might be treatable with a targeted drug therapy that could slow or prevent photoreceptor death, even after the disease process has begun. While not a cure, such a treatment could preserve precious vision.
“This organoid model gave us a clear, human-relevant system to not only understand the ‘why’ but also to start testing the ‘how to fix it,'” the researcher added. “It moves us from observation directly into the preclinical testing phase.”
A New Paradigm for Rare Disease Research
This work on NPHP5-associated LCA is a powerful proof of concept for a new era in medicine. Patient-derived retinal organoids are proving to be indispensable tools for:
- Deciphering Disease Mechanisms: Observing the exact cellular and molecular cascade that leads to vision loss in human tissue.
- Personalized Medicine: Creating organoids from individual patients’ cells to study their specific mutation and predict disease course.
- Drug Screening: Rapidly and ethically testing hundreds of potential therapies in a human system before moving to clinical trials.
For the rare disease community, where patient populations are small and commercial drug development can be challenging, these lab-grown tissues offer an accelerated and more precise path to discovery.
A Ray of Hope
The journey from a lab dish to a clinical treatment is a long one, and the potential drug identified in this study requires much more testing. However, for children and families affected by NPHP5-associated LCA and similar conditions, this research represents a significant beam of light.
By growing a miniature human retina, scientists have uncovered a hidden flaw in the eye’s intricate wiring and found a potential switch to protect it. It’s a profound example of how stem cell technology and lab-grown organs are transforming our fight against blindness, turning fundamental biological insight into tangible hope for restoring sight.